Customizing CoolBox
This document is talking about how to create user-defined Track and Coverage types.
Interactive online version: Binder
CoolBox provided a lot of builtin Track and Coverage types for different kinds of genomic data formats and display form. But maybe these can’t cover all your needs some time.
The good news is that, it’s very easy to define new element types(Track, Coverage, Feature) in CoolBox!
The CoolBox plot system is compitible with matplotlib Python library, you only need to know some basic usage of it.
[1]:
# firstly import the coolbox, and check the version:
import sys; sys.path.insert(0, "../")
import coolbox
from coolbox.api import *
coolbox.__version__
[1]:
'0.3.1'
Custom Track
To implement a new Track, the only three thing you need to do:
Import the base
Trackclass fromcoolbox.core.basemodule.Create new track type, and inherit the
Trackas parent class.Implement the
plotandfetch_data(optional) method.
The code schema will look like this:
from coolbox.core.Track.base import Track
class MyTrack(Track):
def __init__(self, ...):
# Initialization
pass
def plot(self, ax, genome_range, *kwargs):
# Method for plot the track
# within the genome range "chrom:start-end". `genome_range` is a `GenomeRange` object with properties: chrom, start, end
# Draw in the pass-in `ax` Axes
pass
def fetch_data(self, genome_range, **kwargs):
# Method for fetch the data within genome_range
# genome_range is a `coolbox.utilities.GenomeRange` object
pass
For better understand it, here we give an example of two simple toy tracks defination:
Chromosome name track
We can define a simple track to show the chromesome name:
[2]:
from coolbox.core.track.base import Track
from coolbox.utilities import to_gr
import matplotlib.pyplot as plt
class ChromName(Track):
def __init__(self, fontsize=50, offset=0.45):
super().__init__({
"fontsize": fontsize,
"offset": offset,
}) # init Track class
def fetch_data(self, gr, **kwargs):
return gr.chrom # return chromosome name
def plot(self, ax, gr, **kwargs):
x = gr.start + self.properties['offset'] * (gr.end - gr.start)
ax.text(x, 0, gr.chrom, fontsize=self.properties['fontsize'])
ax.set_xlim(gr.start, gr.end)
[3]:
frame = ChromName() + XAxis()
frame.plot("chr9:4000000-6000000")
[3]:
As shown above, we defined a chromesome name display track with just abouot 20 lines of Python code.
Pokemon track
Here we have a more interesting demo:
For example we want to plot some pokemons on some genome regions, and we have a BED like text file like this:
chr1 1000 2000 Bulbasaur
chr1 2000 3000 Charmander
chr1 3000 4000 Squirtle
chr1 4000 5000 Pikachu
We stores it in ./_static/pokemons/list.txt.
[4]:
!ls ./_static/pokemons
Bulbasaur.png Pikachu.png Squirtle.png
Charmander.png PokeBall.png list.txt
[5]:
from coolbox.core.track.base import Track
from coolbox.utilities import to_gr
import pandas as pd
import matplotlib.pyplot as plt
from matplotlib.offsetbox import OffsetImage, AnnotationBbox
class Pokemon(Track):
def __init__(self, file, img_dir):
# read the table
self.df = pd.read_csv(file, sep="\t", header=None)
self.df.columns = ['chrom', 'start', 'end', 'pokemon']
# directory store the images
self.img_dir = img_dir
super().__init__({}) # init Track class
def fetch_data(self, genome_range, **kwargs):
gr = to_gr(genome_range) # force convert to GenomeRange object
df = self.df
sub_df = df[
(df['chrom'] == gr.chrom) & (df['start'] >= gr.start) & (df['end'] <= gr.end)
]
return sub_df
def plot(self, ax, gr, **kwargs):
df = self.fetch_data(f"{gr.chrom}:{gr.start}-{gr.end}") # get data in range
for _, row in df.iterrows():
img_path = f"{self.img_dir}/{row['pokemon']}.png" # get image path
im = plt.imread(img_path)
oi = OffsetImage(im, zoom = 0.5)
px = (row['start'] + row['end']) / 2 # X position
py = 0
box = AnnotationBbox(oi, (px, py), frameon=False)
ax.add_artist(box)
ax.set_xlim(gr.start, gr.end)
ax.set_ylim(-1, 1)
[6]:
pkm = Pokemon("./_static/pokemons/list.txt", "./_static/pokemons/")
frame = XAxis() + Spacer() + pkm
[7]:
# Fetch region data
pkm.fetch_data("chr1:0-6000")
[7]:
| chrom | start | end | pokemon | |
|---|---|---|---|---|
| 0 | chr1 | 1000 | 2000 | Bulbasaur |
| 1 | chr1 | 2000 | 3000 | Charmander |
| 2 | chr1 | 3000 | 4000 | Squirtle |
| 3 | chr1 | 4000 | 5000 | Pikachu |
[8]:
# plot region
frame.plot("chr1:1000-5000")
[8]:

Custom Coverage
Coverage is a layer draw upon the track. Custom Coverage defination is same to the Track, just define the plot and fetch_data (optional) method.
For example we can define a Poke Ball Coverage to draw upon Pokemon Track:
[9]:
from coolbox.core.coverage.base import Coverage
from coolbox.utilities import GenomeRange
pokeball_path = "./_static/pokemons/PokeBall.png"
class PokeBall(Coverage):
def __init__(self, genome_range, img_path=pokeball_path, alpha=0.5):
super().__init__({
"genome_range": GenomeRange(genome_range),
"img_path": img_path,
"alpha": alpha,
})
def plot(self, ax, gr, **kwargs):
gr = self.properties['genome_range']
img_path = self.properties['img_path']
alpha = self.properties['alpha']
im = plt.imread(img_path)
oi = OffsetImage(im, zoom = 0.8, alpha=alpha)
px = (gr.start + gr.end) / 2 # X position
box = AnnotationBbox(oi, (px, 0), frameon=False)
ax.add_artist(box)
[10]:
pkm = Pokemon("./_static/pokemons/list.txt", "./_static/pokemons/")
ball = PokeBall("chr1:1000-2000", alpha=0.6)
frame = XAxis() + Spacer() + pkm + ball
[11]:
frame.plot("chr1:1000-5000")
[11]:

Coverage’s related Track is stored at it’s .track attribute:
[12]:
ball.track
[12]:
<__main__.Pokemon at 0x7fdd82838100>
Maybe you found that track defination is almost same to Track. Actually you can create a Coverage directly from Track class, by using track_to_coverage function, for example, we convert our ChromName Track to Coverage:
[13]:
from coolbox.core.coverage.base import track_to_coverage
ChromNameCov = track_to_coverage(ChromName)
Let’s plot a chromosome name upon pokemon track:
[14]:
pkm = Pokemon("./_static/pokemons/list.txt", "./_static/pokemons/")
frame = XAxis() + Spacer() + pkm + ChromNameCov(offset=0.44)
frame.plot("chr1:1000-5000")
[14]:

Custom Feature
Feature object is used for modify Track or Coverage’s properties (like color, track height, …), for example modify track’s color:
[15]:
bigwig_path = "../../tests/test_data/bigwig_chr9_4000000_6000000.bw"
bw = BigWig(bigwig_path)
frame = XAxis() + bw + Color("#ff0c0c")
frame.plot("chr9:4000000-6000000")
[15]:
We can define our own color Feature:
[16]:
from coolbox.core.feature import Feature
class MyColor(Feature):
def __init__(self, value):
super().__init__(color=value)
[17]:
bw = BigWig(bigwig_path)
frame = XAxis() + bw + MyColor("#0c0cff")
frame.plot("chr9:4000000-6000000")
[17]:
Actually, all properties of CoolBox builtin tracks and coverages is stored in the .properties attribute, it’s a dict object. Add Feature is equivalent to modify this dict:
[18]:
bw.properties
[18]:
{'height': 2,
'color': '#0c0cff',
'name': 'BigWig.2',
'title': '',
'style': 'fill',
'fmt': '-',
'line_width': 2.0,
'size': 10,
'threshold_color': '#ff9c9c',
'threshold': inf,
'cmap': 'bwr',
'alpha': 1.0,
'orientation': None,
'data_range_style': 'y-axis',
'min_value': 'auto',
'max_value': 'auto',
'type': 'line',
'num_bins': 700,
'file': '../../tests/test_data/bigwig_chr9_4000000_6000000.bw'}
[19]:
bw.properties['color'] = "#9c5f2c"
frame = XAxis() + bw
frame.plot("chr9:4000000-6000000")
[19]:
Peakachu
This document shows the ability of cooperating with newly developed algorithms. Peakachu is an machine-learning algorithm used for detecting loops in hic contact matrix.
With the user-friendly plotting and data-fetching api supplied by CoolBox, we can quickly visualize the outputs of Peakachu along with other builtin tracks in CoolBox.
install peakachu
download pretrained model listed in Peakachu
[20]:
!pip install Scikit-learn==0.22 joblib==0.14
!pip install peakachu
Requirement already satisfied: Scikit-learn==0.22 in /Users/bakezq/opt/miniconda3/lib/python3.8/site-packages (0.22)
Requirement already satisfied: joblib==0.14 in /Users/bakezq/opt/miniconda3/lib/python3.8/site-packages (0.14.0)
Requirement already satisfied: scipy>=0.17.0 in /Users/bakezq/opt/miniconda3/lib/python3.8/site-packages (from Scikit-learn==0.22) (1.5.4)
Requirement already satisfied: numpy>=1.11.0 in /Users/bakezq/opt/miniconda3/lib/python3.8/site-packages (from Scikit-learn==0.22) (1.19.4)
Requirement already satisfied: peakachu in /Users/bakezq/opt/miniconda3/lib/python3.8/site-packages (1.1.3)
Define custom track
[21]:
import joblib
import pandas as pd
from scipy.sparse import coo_matrix
from peakachu.scoreUtils import Chromosome
from peakachu import peakacluster
from coolbox.api import *
from coolbox.core.track.hicmat import Cool
from coolbox.utilities import GenomeRange, correspond_track
model = joblib.load('../../tests/test_data/down100.ctcf.pkl')
class Peakachu(Cool):
def __init__(self, *args, **kwargs):
super().__init__(*args, **kwargs)
def fetch_data(self, genome_range1, **kwargs):
matrix = super().fetch_data(genome_range1, **kwargs) # reuse Cool.fetch_data to get the raw contacts
ch = Chromosome(coo_matrix(matrix), model) # call peakachu
pval_ma, raw_ma = ch.score()
self.pval_ma = pval_ma.tocoo()
# should return a np.ndarray representing matrix
return (pval_ma + pval_ma.T).toarray()
@track_to_coverage
class PeakaChuLoops(ArcsBase):
def __init__(self, peakachu, **kwargs):
super().__init__(style="hicpeaks", **kwargs)
peakachu = correspond_track(peakachu)
self.peakachu = peakachu
def fetch_data(self, gr: GenomeRange, **kwargs):
ma = self.peakachu.pval_ma
mask = ma.data > 0.87 # threshold to filter pvals
pixels = dict(zip(zip(ma.row[mask], ma.col[mask]), 1 / ma.data[mask]))
rows = [(x, x + 1, y, y + 1) # call peakachu
for [x, y], *_ in peakacluster.local_clustering(pixels, res=10000)]
# should return a pd.DataFrame with columns ['start1', 'end1', 'start2', 'end2']
peaks = pd.DataFrame(rows, columns=['start1', 'end1', 'start2', 'end2']) * self.peakachu.fetched_binsize + gr.start
return peaks
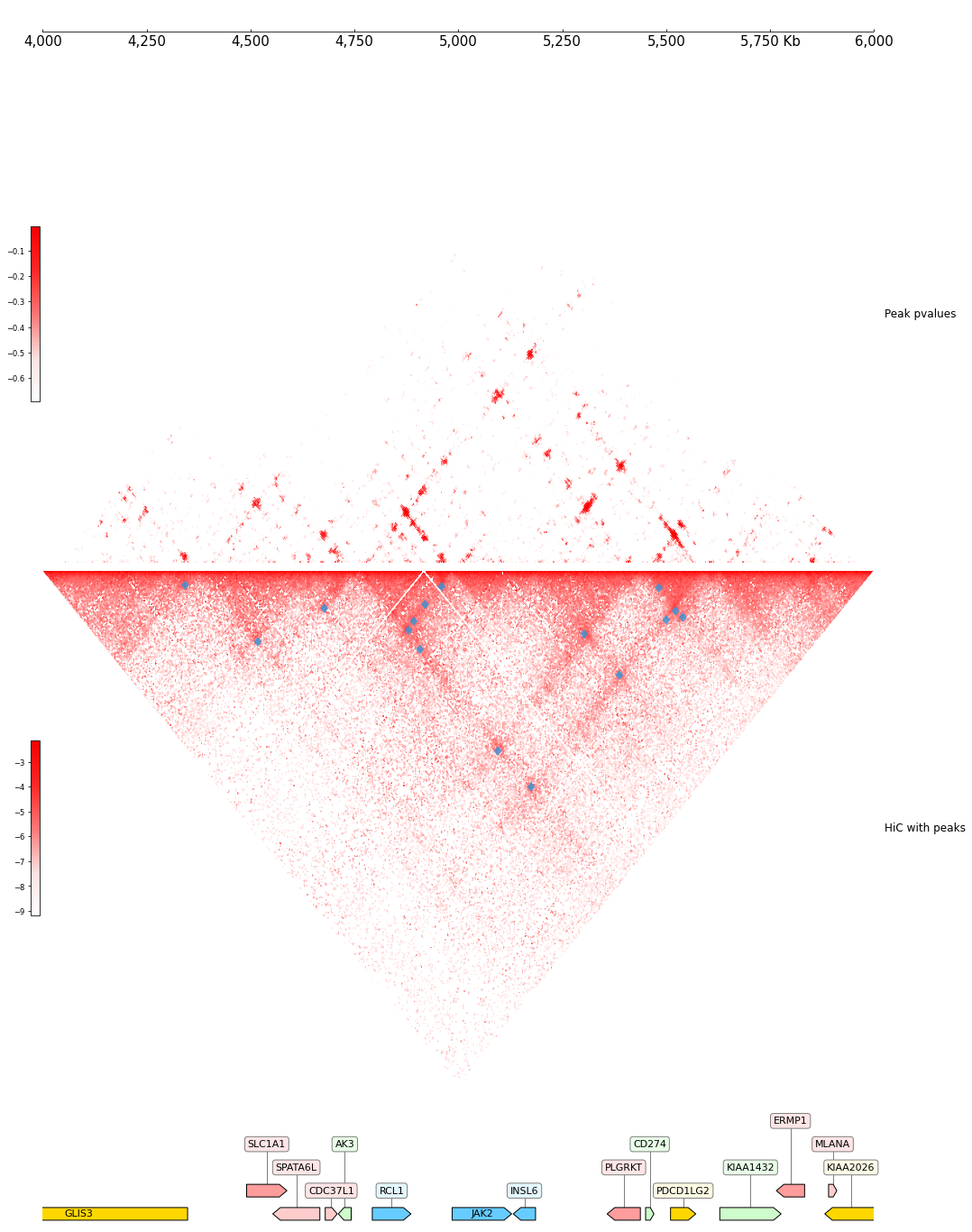
Visualize Track
[22]:
DATA_DIR = "../../tests/test_data"
TEST_RANGE = "chr9:4000000-6000000"
RANGE_MARK = "chr9_4000000_6000000"
COOL_PATH = f"{DATA_DIR}/cool_{RANGE_MARK}.mcool"
PVALS = Peakachu(COOL_PATH, style="triangular")
PEAKS = Cool(COOL_PATH, style="triangular", orientation="inverted") + \
PeakaChuLoops(PVALS, line_width=5)
frame = XAxis() + \
PVALS + Title("Peak pvalues") + \
PEAKS + Title("HiC with peaks") + \
Spacer(0.5) + \
GTF(f"{DATA_DIR}/gtf_{RANGE_MARK}.gtf") + TrackHeight(5)
frame.plot(TEST_RANGE)
scoring matrix chrm
num candidates 79800
[22]:
CLI Mode
Custom elements can also used in CLI mode, just save your definition to a .py file, and load_module in CLI:
[23]:
%%bash
module_path=./_static/peakachu_track.py
data_dir=../../tests/test_data/
cool_path=$data_dir/cool_chr9_4000000_6000000.mcool
gtf_path=$data_dir/gtf_chr9_4000000_6000000.gtf
coolbox load_module $module_path - \
add XAxis - \
add Peakachu $cool_path --style "triangular" --name "pvals" - \
add Cool $cool_path --style "triangular" --orientation "inverted" - \
add PeakaChuLoops "pvals" --line_width 5 -\
add GTF $gtf_path --height 5 - \
goto "chr9:4000000-6000000" - \
plot "/tmp/test_coolbox_peakachu.pdf"
scoring matrix chrm
num candidates 79800
Browser Mode
Custom elements are naturally adaptive to browser mode. For use custom tracks defined above, just run:
[24]:
bsr = Browser(frame)
bsr.goto(TEST_RANGE)
bsr.show()
scoring matrix chrm
num candidates 80200